Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.biortech.2011.02.026.
References
Aisaka, K., Igarashi, A., Yamaguchi, K., Uwajima, T., 1991. Purification, crystallization and characterization of N-acetylneuraminate lyase from Escherichia coli. Biochem. J. 276, 541–546.
Aytar, B.S., Bakir, U., 2008. Preparation of cross-linked tyrosinase aggregates. Process. Biochem. 43, 125–131.
Blayer, S., Woodley, J.M., Lilly, M.D., 1996. Characterization of the chemoenzymatic synthesis of N-acetyl-D-neuraminic acid (Neu5Ac). Biotechnol. Prog. 12, 758– 763.
Cabana, H., Jones, J.P., Agathos, S.N., 2007. Preparation and characterization of cross- linked laccase aggregates and their application to the elimination of endocrine disrupting chemicals. J. Biotechnol. 132, 23–31.
Cabirol, F.L., Tan, P.L., Tay, B., Cheng, S., Hanefekd, U., Sheldon, R.A., 2008. Linum usitatissium hydroxynitrile lyse cross-linked enzyme aggregates: a recyclable enantioselective catalyst. Adv. Synth. Catal. 350, 2329–2338.
Cao, L., van Rantwijk, F., Sheldon, R.A., 2000. Cross-linked enzyme aggregates: a simple and effective method for the immobilization of penicillin acylase. Org. Lett. 2, 1361–1364.
Dalal, S., Kapoor, M., Grupta, M.N., 2007. Preparation and characterization of combi- CLEAs catalyzing multiple non-cascade reactions. J. Mol. Catal. B: Enzyme 44, 128–132.
Dong, T., Zhao, L., Huang, Y., Tan, X., 2010. Preparation of cross-linked aggregates of aminoacylase from Aspergillus melleus by using bovine serum albumin as an inert additive. Bioresour. Technol. 101, 6569–6571.
Gupta, P., Dutt, K., Misra, S., Raghuwanshi, S., Saxena, R.K., 2009. Characterization of cross-linked immobilized lipase from thermophilic mould Thermomyces lanuginosa using glutaraldehyde. Bioresour. Technol. 100, 4074–4076.
Hara, P., Hanefeld, U., Kanerva, L.T., 2008. Sol–gels and cross-linked aggregates of lipase PS from Burkholderia cepacia and their application in dry organic solvents. J. Mol. Catal. B: Enzyme 50, 80–86.
Kragl, U., Gygax, D., Ghisalba, O., Wandrey, C., 1991. Enzymatic two-step synthesis of N-acetyl-neuraminic acid in the enzyme membrane reactor. Angew. Chem. Int. Ed. Engl. 30, 827–828.
Kyte, J., Doolittle, R.F., 1982. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157, 105–132.
Liese, A., Seelbach, K., Buchholz, A., Haberland, J., 2006. Processes. In: Liese, A., Seelbach, K., Wandrey, C. (Eds.), Industrial Biotransformations. Wiley-VCH, Weinheim, Germany, pp. 457–460.
Mahmoudian, M., Noble, D., Drake, C.S., Middleton, R.F., Montgomery, D.S., Piercey, J.E., Ramlakhan, D., Todd, M., Dawson, M.J., 1997. An efficient process for production of N-acetylneuraminic acid using N-acetylneuraminic acid aldolase. Enzyme Microbiol. Technol. 20, 393–400.
Mateo, C., Palomo, J.M., van Langen, L.M., van Rantwijk, F., Sheldon, R.A., 2004. A new, mild crosslinking methodology to prepare crosslinked enzyme aggregates. Biotechnol. Bioeng. 86, 273–276.
Matijosyte, I., Arends, I.W.C.E., de Vries, S., Sheldon, R.A., 2010. Preparation and use of cross-linked enzyme aggregates (CLEAs) of laccases. J. Mol. Catal. B: Enzyme 62, 142–148.
Montoro-García, S., Gil-Ortiz, F., Navarro-Fernández, J., Rubio, V., García-Carmona, F., Sánchez-Ferrer, A., 2010. Improved cross-linked enzyme aggregates for the production of desacetyl b-lactam antibiotics intermediates. Bioresour. Technol. 101, 331–336.
Müller, F.M., Werner, K.E., Kasai, M., Francesconi, A., Chanock, S.J., Walsh, T.J., 1998. Rapid extraction of genomic DNA from medically important yeasts and filamentous fungi by high-speed cell disruption. J. Clin. Microbiol. 6, 1625– 1629.
Nahàlka, J., Vikartovská, A., Hrabárová, E., 2008. A crosslinked inclusion body process for sialic acid synthesis. J. Biotechnol. 134, 146–153.
Roessl, U., Nahálka, J., Nidetzky, B., 2010. Carrier-free immobilized enzymes for biocatalysis. Biotechnol. Lett. 32, 341–350.
Sánchez-Carrón, G., García-García, M.I., López-Rodríguez, A.B., Jiménez-García, S., Sola-Carvajal, A., García-Carmona, F., Sánchez-Ferrer, A., 2011. Molecular characterization of a novel N-acetylneuraminate lyase from Lactobacillus plantarum WCFS1. Appl. Envirom. Microbiol, doi:10.1128/AEM.02927-10 [Epub ahead of print].
Sangeetha, K., Abraham, T.E., 2008. Preparation and characterization of cross-linked enzyme aggregates (CLEAs) of subtilisin for controlled release applications. Int. J. Biol. Macromol. 43, 314–319.
Schoevaart, R., Wolbers, M.W., Golubovic, M., Ottens, M., Kieboom, A.P.G., van Rantwijk, F., van der Wielen, L.A.M., Sheldon, R.A., 2004. Preparation, optimization, and structures of cross-linked enzyme aggregates (CLEAs). Biotechnol. Bioeng. 87, 754–762.
Shah, S., Sharma, A., Gupta, M.N., 2006. Preparation of cross-linked enzyme aggregates by using bovine serum albumin as a proteic feeder. Anal. Biochem. 351, 207–213.
Sheldon, R.A., 2007. Cross-linked enzyme aggregates (CLEAÒs): stable and recyclable biocatalysts. Biochem. Soc. Trans. 35, 1583–1587.
Sheldon, R.A., 2010. Cross-linked enzyme aggregates as industrial biocatalysts. In: Shioiri, T., Izawa, K., Konoike, T. (Eds.), Pharmaceutical Process Chemistry. Wiley-VCH, Weinheim, Germany. doi:10.1002/9783527633678.ch8.
Sugai, T., Kuboki, A., Hiramatsu, S., Okazaki, H., Ohta, H., 1995. Improved enzymatic procedure for a preparative-scale synthesis of sialic acid and KDN. Bull. Chem. Soc. Jpn. 68, 3581–3589.
Tyagi, R., Batra, R., Grupta, M.N., 1999. Amorphous enzyme aggregates: stability towards heat and aqueous-organic cosolvents mixtures. Enzyme Microbiol. Technol. 24, 348–353.
van Langen, L.M., Selassa, R.P., van Rantwijk, F., Sheldon, R.A., 2005. Cross-linked aggregates of (R)-oxynitrilase: a stable, recyclable biocatalyst for enantioselective hydrocyanation. Org. Lett. 7, 327–329.
Wang, M., Qi, W., Yu, Q., Su, R., He, Z., 2010. Cross-linking enzyme aggregates in the macropores of silica gel: a practical and efficient method for enzyme stabilization. Biochem. Eng. J. 54, 168–174.
Wang, M., Jia, C., Qi, W., Yu, Q., Peng, X., Su, R., He, Z., 2011. Porous-CLEAs of papain: application to enzymatic hydrolysis of macromolecules. Bioresour. Technol. 102, 3541–3545.
Wilson, L., Betancor, L., Fernandez-Lorente, G., Fuentes, M., Hidalgo, A., Guisan, J.M., Pessela, B.C.C., Fernandez-Lafuente, R., 2004. Crosslinked aggregates of multimeric enzymes: a simple and efficient methodology to stabilize their quaternary structure. Biomacromolecules 5, 814–817.
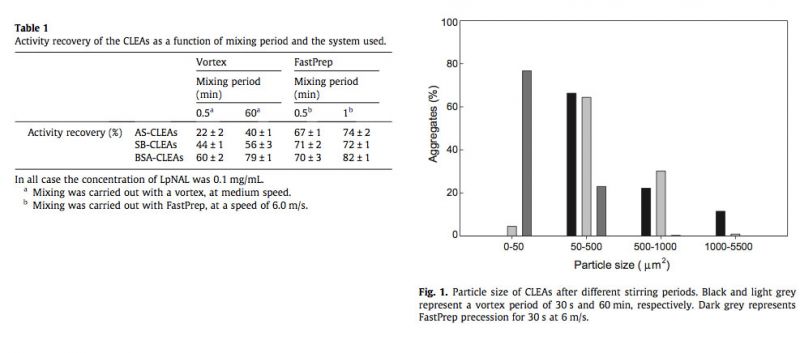
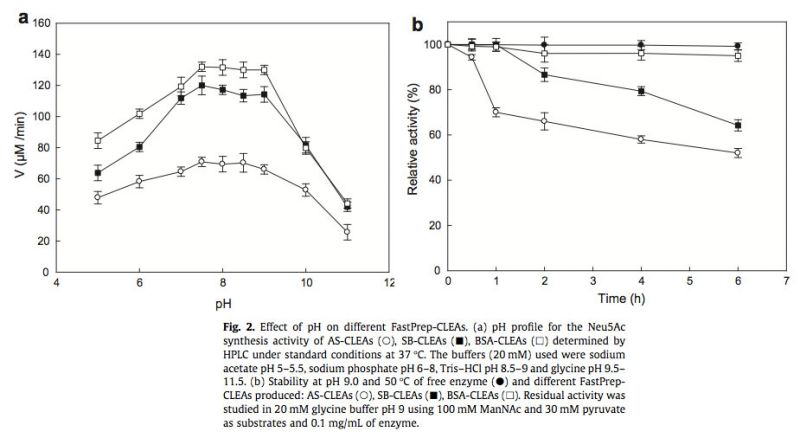
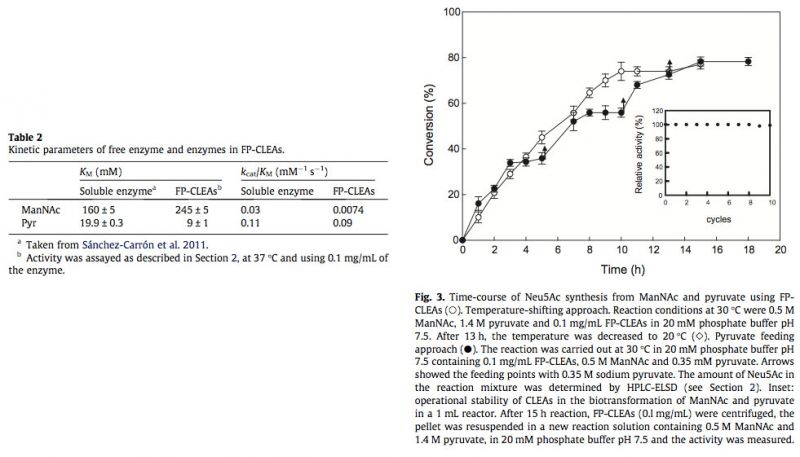 3.4. Potential production of Neu5Ac
3.4. Potential production of Neu5Ac


