Delphine Meynard1,2,4, Léon Kautz1,2,4, Valérie Darnaud1,2, Franc ̧ois Canonne-Hergaux3, Hélène Coppin1,2 & Marie-Paule Roth1,2
Expression of hepcidin, a key regulator of intestinal iron absorption, can be induced in vitro by several bone morphogenetic proteins (BMPs), including BMP2, BMP4 and BMP9 (refs. 1,2). However, in contrast to BMP6, expression of other BMPs is not regulated at the mRNA level by iron in vivo3, and their relevance to iron homeostasis is unclear. We show here that targeted disruption of Bmp6 in mice causes a rapid and massive accumulation of iron in the liver, the acinar cells of the exocrine pancreas, the heart and the renal convoluted tubules. Despite their severe iron overload, the livers of Bmp6-deficient mice have low levels of phosphorylated Smad1, Smad5 and Smad8, and these Smads are not significantly translocated to the nucleus. In addition, hepcidin synthesis is markedly reduced. This indicates that Bmp6 is critical for iron homeostasis and that it is functionally nonredundant with other members of the Bmp subfamily. Notably, Bmp6-deficient mice retain their capacity to induce hepcidin in response to inflammation. The iron burden in Bmp6 mutant mice is significantly greater than that in mice deficient in the gene associated with classical hemochromatosis (Hfe), suggesting that mutations in BMP6 might cause iron overload in humans with severe juvenile hemochromatosis for which the genetic basis has not yet been characterized.
Hepcidin, a key regulator of iron absorption, binds to the cellular iron exporter ferroportin and induces its endocytosis and proteolysis, preventing release of iron from macrophages or intestinal cells into the plasma(4). In genetic hemochromatosis, sustained deficiency of hepcidin(5–8) causes excessive iron absorption from the diet and leads to the deposition of iron in the liver and other tissues, with consequent organ damage and functional failure. Hepcidin expression is controlled by the BMP signaling pathway(1,9). The signal is initiated when BMP ligands bind and activate receptor serine-threonine kinases at the hepatocyte cell surface. The resulting receptor complex propagates the signal through phosphorylation of cytoplasmic effectors, the receptor- regulated Smad1, Smad5 and Smad8. Once phosphorylated, the Smad proteins 1, 5 and 8 form heteromeric complexes with the common mediator Smad4 and translocate to the nucleus, where they modulate transcription of target genes10. Although many BMP ligands, including BMP2, BMP4 and BMP9, can positively regulate hepcidin expression in vitro(2,9,11), it is not yet known how BMP signaling is modulated in response to body iron status or which BMPs are the endogenous regulators of hepcidin expression and iron homeostasis in vivo.

A functional genomic study in mice fed either an iron-enriched or an iron-deficient diet allowed us to determine that, in contrast to that of other Bmp genes, Bmp6 mRNA expression was regulated by iron similarly to Hamp (hepcidin) mRNA expression, and suggested that Bmp6 has a preponderant role in the activation of the Smad signaling pathway leading to hepcidin synthesis in vivo(3). Notably, Bmp6 is expressed in hepatocytes, and its abundance, as detected by immunochemistry, strongly increases in wild-type mice fed an iron-enriched diet (data not shown). We therefore suspected that iron homeostasis would be disturbed in a strain of Bmp6-deficient mice derived ten years ago(12). In contrast to mice deficient for other Bmp molecules, these mice are viable and fertile. Although Bmp6 mutant embryos show a delay in ossification, strictly confined to the developing sternum, newborn and adult Bmp6 mutants have skeletal elements indistinguishable from those of wild-type mice, implying that Bmp6 is not required for normal skeletal development.
In agreement with our expectations, we found that targeted disruption of Bmp6 results in massive iron overload. At 7 weeks of age, serum transferrin saturations in Bmp6 -/- mice were close to 100%. The mice also had 11-fold more non-heme liver iron than had wild-type mice, whereas their splenic non-heme iron content was lower than that in wild-type controls (Fig. 1). We further examined the sites of iron accumulation by staining histological sections for iron. In Bmp6 -/- mice at 7 weeks of age, there was considerable iron accumulation in liver parenchymal cells (hepatocytes) but minimal iron staining in splenic macrophages. Hepatocellular iron deposition was extending from periportal to centrilobular hepatocytes, following blood flow in the liver (Fig. 2). Iron accumulation was also observed in acinar cells of the exocrine pancreas, in the heart (see Supplementary Fig. 1 online) and in the renal convoluted tubules.
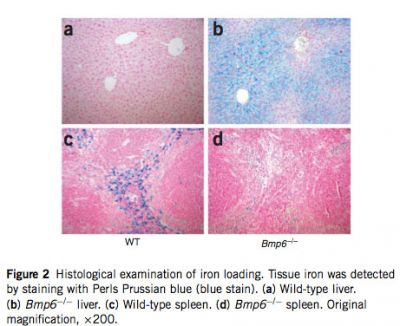 |
 |
By 7 weeks of age, Bmp6 -/- mice have accumulated significantly more iron than 12-week-old mice of different genetic backgrounds deficient for the classical hemochromatosis gene Hfe (M.P. Roth, unpublished data). Targeted disruption of the mouse Bmp6 gene thus results in a severe iron-loading phenotype similar to that of mice deficient for Hfe2 (also known as Hjv), which encodes hemo- juvelin(13,14), for Smad4 (ref. 11), or for Hamp(15), and to that of humans with juvenile hemochromatosis(16). We conclude that Bmp6 is critical in the control of iron homeostasis. We next examined liver mRNA expression of hepcidin and other genes previously shown to be regulated by iron similarly to hepcidin. We found that Bmp6 -/- mice had approximately 22-fold less hepatic Hamp mRNA than wild-type controls (Fig. 1). Notably, expression of other genes known (Id1, Smad7) or suspected (Atoh8) to be targets of the BMP-Smad signaling pathway(3) was markedly reduced in the liver of Bmp6 -/- mice (see Supplementary Fig. 2 online).
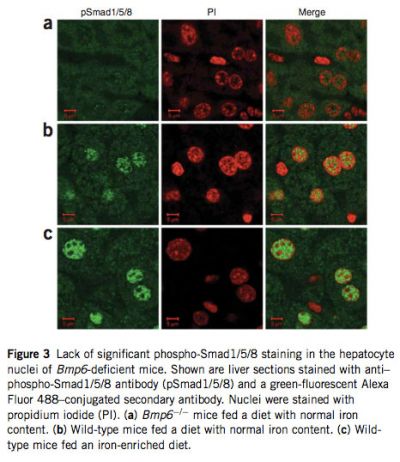
Because Bmp6 transmits signal through phosphorylation of Smad1, Smad5 and Smad8, we compared the relative abundance of phos- phorylated forms of these three Smads in liver extracts of Bmp6 -/- and wild-type mice by protein blot analysis. As expected, phosphorylation of all three was significantly reduced in Bmp6 -/- mice (see Supplementary Fig. 3 online). We then examined nuclear translocation of these three Smads by labeling liver tissue sections with an antibody that detects Smad1, Smad5 and Smad8 when phosphorylated at serines in the C-terminal domain. In Bmp6 -/- mice, immunostaining of phospho-Smad1/5/8 was weak and distributed evenly in cytoplasm and nucleus, confirming that levels of phosphorylation of these Smads were insignificant and explaining why Hamp mRNA levels are markedly reduced in these mice. In contrast, phospho- Smad1/5/8 staining was observed in the hepatocyte nuclei of wild-type mice fed a diet with normal iron content and was strongly induced in those of wild-type animals fed an iron-enriched diet for 1 week to induce iron overload (Fig. 3).
|
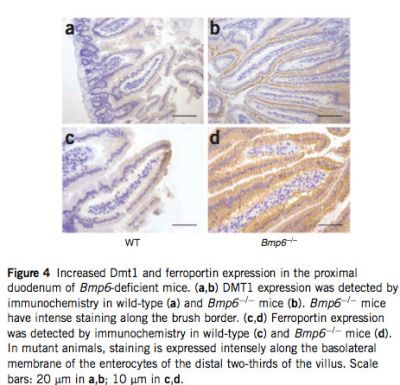
The marked iron accumulation in Bmp6-deficient mice led us to evaluate the expression of genes involved in duodenal iron absorption by real-time quantitative PCR. Transcript levels of the genes encoding the brush-border surface ferric reductase Dcytb (Cybrd1) and the apical transmembrane iron transporter Dmt1 (Slc11a2) were elevated about 14.6- and 13.8-fold, respectively, whereas that of the gene encoding the basolateral membrane transporter ferroportin (Slc40a1) was increased about 2.2-fold (see Supplementary Fig. 4 online). This increase may be induced by the smaller amount of stainable iron present in proximal duodenal enterocytes in Bmp6 -/- mice compared with wild-type controls (data not shown). The induction of Dmt1 in the duodenum was confirmed by immunohistochemistry: weak staining of Dmt1 protein was detectable in wild-type controls, but Bmp6-/- mice had intense staining along the brush border (Fig. 4). We also examined ferroportin expression by immunohistochemistry, focusing on tissues in which ferroportin is known to be important. Whereas ferroportin is normally expressed at low levels in the absorptive enterocytes lining the intestinal villi, in Bmp6-/- mice we observed a massive increase in ferroportin protein expressed at the basolateral membrane (Fig. 4). Similarly, ferroportin expression was markedly enhanced in tissue macrophages of the livers and spleens of Bmp6-/- mice compared with those of wild-type controls (see Supplementary Fig. 5 online). This is consistent with a lack of hepcidin expression in these mice, leading to stabilization of ferroportin at the membrane of enterocytes and tissue macrophages.
|
We next investigated the possible role of Bmp6 in the induction of hepcidin by inflammation. Hepcidin is part of the type II acute-phase response and is thought to have a crucial role in anemia of chronic disease. Whereas hepcidin is induced by activation of the inflammatory pathway in Hfe2-deficient mice(14), this induction is not observed in mice with liver-specific Smad4 deficiency(11). We therefore set out to determine whether lipopolysaccharide (LPS)-dependent induction of hepcidin requires Bmp6 by treating Bmp6 mutant mice and wild-type controls with LPS or saline solution. As expected, the acute-phase genes Il6, Tnf and Crp were strongly induced in LPS-treated mice as compared with saline-treated animals (see Supplementary Figs. 6 and 7 online). Hamp gene expression was induced about 24-fold in response to the LPS treatment in Bmp6 -/- mice and 2.6-fold in wild-type controls (Fig. 1). Notably, Hamp mRNA levels in LPS- injected Bmp6 -/- animals did not reach those of wild-type controls. These findings suggest that the total level of Hamp mRNA observed upon inflammation is additive to the baseline level and argue for the existence of two independent pathways that lead to the regulation of hepcidin expression. Of these two pathways, only the iron-sensing pathway requires functional Bmp6 and hemojuvelin(14). It is not clear yet how LPS activates hepcidin production in Bmp6-deficient mice, although there is evidence supporting a role for BMP–transforming growth factor β (TGF-β) signaling. Indeed, transcriptional activation of hepcidin by interleukin-6 (IL-6) is abrogated in mice with liver-specific conditional knockout of Smad4 (ref. 11), and chemical inhibition of BMP signal transduction in a human hepatoma cell line blocks the induction of hepcidin expression not only by BMPs but also by IL-6 (ref. 17). Furthermore, a BMP-responsive element in the Hamp promoter is required to control hepatic expression in response to IL-6 (ref. 18). Further work is needed to identify which TGF-β–BMP superfamily ligands, other than BMP6, function as endogenous activators of hepcidin expression during inflammation.
BMP molecules were initially identified by their capacity to induce endochondral bone formation. However, mild and/or extremely localized skeletal defects are observed in mice deficient for Bmp1, Bmp2, Bmp4 and Bmp7, which contrasts strongly with the profound and specific effects seen on gut, heart, neural tube or kidney morphogenesis. The physiological actions of BMPs in soft tissues thus appear more important than their actions in the skeleton(19,20). Bmp6 -/- mice are notable for their lack of skeletal defects but, curiously, no effect of Bmp6 deficiency on other tissues or organs has been reported so far. Our data show a previously unsuspected but essential role of Bmp6 in the maintenance of iron homeostasis. Although other Bmp molecules are functional in the severely iron-overloaded Bmp6-deficient mice, they do not compensate for the absence of Bmp6, demonstrating that this ironregulatory function is unique to Bmp6 among the BMP family. Future studies designed to establish how iron regulates Bmp6 expression will be useful to fully understand regulation of hepcidin synthesis by iron. The iron burden in Bmp6 mutant mice is significantly more severe than that in Hfe-deficient mice and closely resembles that seen in Hfe2-, Hamp- or Smad4-deficient mice. This suggests that the human BMP6 gene could be a candidate locus in people with severe juvenile hemochromatosis not attributable to hemojuvelin or hepcidin. Individuals with mutations in HFE, in the gene encoding transferrin receptor 2 (TFR2) and in HFE2 have low hepcidin(5,6,21) and are consequently unable to effectively repress iron absorption. Although the mechanisms by which transferrin receptor 2 and HFE regulate hepcidin remain enigmatic, hemojuvelin has been shown to be a cell surface BMP co-receptor and to augment signal transduction through this pathway(9). Notably, although soluble hemojuvelin inhibits induction of hepcidin by several BMP ligands in vitro, careful examination of the data shows that this inhibition is far more efficient for BMP6 (ref. 1). These results suggest that hemojuvelin is a co-receptor for BMP6 in vivo and that BMP6 and hemojuvelin act coordinately to induce hepcidin expression.
METHODS
Mice. Bmp6-null mice (Bmp6 m1Rob) were derived as previously described(12) and maintained on a background derived from the same stock (outbred CD1) as wild-type controls. The targeted allele was confirmed to encode a loss-of- function mutation. We checked that insertion of the neomycin-resistance selection marker in exon 2 of Bmp6 had not caused deficiency of Txndc5, a gene adjacent to Bmp6, in the liver of these mice (data not shown). All experiments were performed on males. Unless otherwise specified, mice received a diet with normal iron content (200 mg iron per kg body weight; SAFE) and were analyzed at 7 weeks and fasted for 14 h before they were killed. Experimental iron overload was obtained by feeding wild-type controls an iron- enriched diet (8.5 g per kg). Experimental protocols were approved by the Midi-Pyrénées Animal Ethics Committee.
LPS injection. LPS (1 mg per g body weight; serotype 055:B5; Sigma) or an equivalent volume of saline solution (0.9% NaCl) was injected intraperitoneally, and organs (liver and spleen) were isolated 6 h after injection.
Tissue iron staining and quantitative tissue iron measurement. Intestine, liver, spleen, heart, pancreas and kidney samples were fixed in 10% buffered formalin and embedded in paraffin. Deparaffinized tissue sections were stained with the Perls Prussian blue stain for non-heme iron and counterstained with nuclear fast red. Quantitative measurement of non-heme iron in the liver and the spleen was performed as described previously(22). Results are reported as mg of iron per g wet weight of tissue.
Immunohistochemistry. Four-micrometer sections of paraffin-embedded tissues were mounted on glass slides. Endogenous peroxidase activity was quenched by incubating specimens with Peroxidase Block (DakoCytomation EnVision+ System-HRP, Dako). Sections were blocked in PBS containing 1% BSA and 10% FCS (Invitrogen) and incubated for 1 h at 20°C with primary antibody to DMT1 (anti-DMT1; ref. 23) or for 1 h 30 min at 37°C with primary anti-ferroportin(24) diluted in PBS/1% BSA. Immunohistochemical staining was performed using the DakoCytomation EnVision+ System-HRP according to the manufacturer’s instructions. Sections were counterstained with hematoxylin.
Immunofluorescence. Nonspecific fluorescence due to endogenous avidin and biotin was blocked by the Avidin Biotin Blocking Solution (Lab Vision). After permeabilization with 0.3% Triton X-100 in PBS/5% BSA, liver tissue sections were incubated overnight at 4°C with a rabbit polyclonal antibody to phosphorylated Smad1, Smad5 and Smad8 (polyclonal anti–phospho- Smad1/5/8; 1/100; Cell Signaling Technology). Staining was obtained using the secondary Alexa Fluor 488–goat anti-rabbit (1/200; Invitrogen), and slides were mounted in Vectashield mounting medium containing propidium iodide (Clinisciences) to counterstain DNA. Cells were visualized using a Zeiss LSM 510 confocal fluorescent microscope with an x63 oil-immersion objective.
RNA preparation and real-time quantitative PCR. Liver, spleen and duodenum samples were dissected for RNA isolation, rapidly frozen and stored in liquid nitrogen. Total RNA was extracted and purified using the RNeasy Lipid Tissue kit (Qiagen). All primers were designed using the Primer Express 2.0 software (Applied Biosystems) and are detailed in Supplementary Table 1 online. Real-time quantitative PCR (Q-PCR) reactions were prepared with M-MLV reverse transcriptase (Promega) and LightCycler 480 DNA SYBR Green I Master reaction mix (Roche Diagnostics) as previously described(22) and run in duplicate on a LightCycler 480 Instrument (Roche Diagnostics).
Protein blot analysis. Livers were homogenized in a FastPrep-24 Instrument (MP Biomedicals Europe) for 20 s at 4 m s–1. The lysis buffer (50 mM Tris-HCl, pH 8, 150 mM NaCl, 5 mM EDTA, pH 8, 1% NP-40) included inhibitors of proteases (1 mM PMSF, 10 mg ml–1 leupeptin, 10 mg /ml–1 pepstatin A and 1 mg ml–1 antipain) and of phosphatases (10 ml ml–1 phosphatase inhibitor cocktail 2, Sigma-Aldrich). Proteins were quantified using the Bio-Rad Protein Assay kit based on the Bradford method. Protein extracts (30 mg for phospho- Smad1/5/8 and 60 mg for Smad5) were diluted in Laemmli buffer (Sigma-Aldrich), incubated for 5 min at 95 1C and subjected to SDS-PAGE. Proteins were then transferred to Hybond-C Extra nitrocellulose membranes (Amersham Biosciences). Membranes were blocked with Odyssey blocking buffer (LI-COR Biosciences), incubated with rabbit polyclonal anti–phospho- Smad1/5/8 (1/500, Cell Signaling Technology) or a goat polyclonal anti-Smad5 (1/200, Santa Cruz Biotechnology) and a mouse monoclonal anti–b-actin (1/20,000, Sigma-Aldrich) at 4°C overnight and washed with PBS/0.1% Tween-20 buffer. After incubation with infrared IRDye 800 anti-rabbit or anti-goat and secondary IRDye 680 goat anti-mouse (1/15,000, LI-COR Biosciences), membranes were scanned on the Odyssey Infrared Imaging System. Band sizing was performed using the Odyssey 3.0 software (LI-COR Biosciences), and quantification of phospho-Smad1/5/8 activity and of Smad5 was calculated by normalizing the specific probe band to β-actin.
Statistical analyses. Log-transformed values of liver and spleen iron contents were compared by Student’s t-tests. The relative expression ratios (and s.e.m. values) for liver, spleen and/or duodenum transcripts between Bmp6 -/- mice and wild-type controls or LPS- and saline-treated animals were calculated using the relative expression software tool (REST, http://rest.gene-quantification. info)(25). The mathematical model is based on the mean crossing point (Cp) deviation between sample and control groups of target genes, normalized by the mean Cp deviation of the reference gene Gusb (encoding β-glucuronidase)(26). An efficiency correction was performed and randomization tests, which have the advantage of making no distributional assumptions about the data, were used to determine statistical significance.
Note: Supplementary information is available on the Nature Genetics website.
ACKNOWLEDGMENTS
The authors thank E. Robertson (Dunn School of Pathology, University of Oxford) for kindly providing the Bmp6-deficient mice, C. Rouanet for excellent technical assistance, J. Seumois and M. Calise (Service de Zootechnie, IFR30, Toulouse) for their help with the mouse breeding, and S. Allart (Cellular Imaging platform, IFR30), F. Capilla and T. Al Saati (Experimental Histopathology platform, IFR30) for skilled advice. This work was supported in part by grants from the Agence Nationale pour la Recherche (ANR, programme IRONGENES), the European Commission (LSHM-CT-2006-037296: EUROIRON1) and the Fondation pour la Recherche Médicale (to L.K.).
AUTHOR CONTRIBUTIONS
D.M., L.K. and V.D. performed phenotype assessment, analyzed data and reviewed the paper; F.C.-H. provided antibodies and technical advice; H.C. and M.-P.R. conceived the project and wrote the manuscript. The last two senior authors contributed equally to the work.
Published online at http://www.nature.com/naturegenetics/
Reprints and permissions information is available online at http://npg.nature.com/ reprintsandpermissions/
1. Babitt, J.L. et al. Modulation of bone morphogenetic protein signaling in vivo regulates systemic iron balance. J. Clin. Invest. 117, 1933–1939 (2007).
2. Truksa, J., Peng, H., Lee, P. & Beutler, E. Bone morphogenetic proteins 2, 4, and 9 stimulate murine hepcidin 1 expression independently of Hfe, transferrin receptor 2 (Tfr2), and IL-6. Proc. Natl. Acad. Sci. USA 103, 10289–10293 (2006).
3. Kautz, L. et al. Iron regulates phosphorylation of Smad1/5/8 and gene expression of Bmp6, Smad7, Id1, and Atoh8 in the mouse liver. Blood 112, 1503–1509 (2008).
4. Nemeth, E. et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 306, 2090–2093 (2004).
5. Nemeth, E., Roetto, A., Garozzo, G., Ganz, T. & Camaschella, C. Hepcidin is decreased in TFR2 hemochromatosis. Blood 105, 1803–1806 (2005).
6. Papanikolaou, G. et al. Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis. Nat. Genet. 36, 77–82 (2004).
7. Piperno, A. et al. Blunted hepcidin response to oral iron challenge in HFE-related hemochromatosis. Blood 110, 4096–4100 (2007).
8. Roetto, A. et al. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat. Genet. 33, 21–22 (2003).
9. Babitt, J.L. et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat. Genet. 38, 531–539 (2006).
10. Shi, Y. & Massague, J. Mechanisms of TGF-b signaling from cell membrane to the nucleus. Cell 113, 685–700 (2003).
11. Wang, R.H. et al. A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab. 2, 399–409 (2005).
12. Solloway, M.J. et al. Mice lacking Bmp6 function. Dev. Genet. 22, 321–339 (1998).
13. Huang, F.W., Pinkus, J.L., Pinkus, G.S., Fleming, M.D. & Andrews, N.C. A mouse model of juvenile hemochromatosis. J. Clin. Invest. 115, 2187–2191 (2005).
14. Niederkofler, V., Salie, R. & Arber, S. Hemojuvelin is essential for dietary iron sensing, and its mutation leads to severe iron overload. J. Clin. Invest. 115, 2180–2186 (2005).
15. Lesbordes-Brion, J.C. et al. Targeted disruption of the hepcidin 1 gene results in severe hemochromatosis. Blood 108, 1402–1405 (2006).
16. Cox, T.M. & Halsall, D.J. Hemochromatosis—neonatal and young subjects. Blood Cells Mol. Dis. 29, 411–417 (2002).
17. Yu, P.B. et al. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat. Chem. Biol. 4, 33–41 (2008).
18. Verga Falzacappa, M.V., Casanovas, G., Hentze, M.W. & Muckenthaler, M.U. A bone morphogenetic protein (BMP)-responsive element in the hepcidin promoter controls HFE2-mediated hepatic hepcidin expression and its response to IL-6 in cultured cells. J. Mol. Med. 86, 531–540 (2008).
19. Ducy, P. & Karsenty, G. The family of bone morphogenetic proteins. Kidney Int. 57, 2207–2214 (2000).
20. Gazzerro, E. & Canalis, E. Bone morphogenetic proteins and their antagonists. Rev. Endocr. Metab. Disord. 7, 51–65 (2006).
21. Bridle, K.R. et al. Disrupted hepcidin regulation in HFE-associated haemochromatosis and the liver as a regulator of body iron homoeostasis. Lancet 361, 669–673 (2003).
22. Dupic, F. et al. Inactivation of the hemochromatosis gene differentially regulates duodenal expression of iron-related mRNAs between mouse strains. Gastroenterology 122, 745–751 (2002).
23. Canonne-Hergaux, F. et al. Expression of the DMT1 (NRAMP2/DCT1) iron transporter in mice with genetic iron overload disorders. Blood 97, 1138–1140 (2001).
24. Delaby, C., Pilard, N., Goncalves, A.S., Beaumont, C. & Canonne-Hergaux, F. Presence of the iron exporter ferroportin at the plasma membrane of macrophages is enhanced by iron loading and down-regulated by hepcidin. Blood 106, 3979–3984 (2005).
25. Pfaffl, M.W., Horgan, G.W. & Dempfle, L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 30, e36 (2002).
26. Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, e45 (2001).


