Medical Mycology August 2009, 47, 508-514
JEAN-FRANC ̧ OIS DURANT*, PIERRE-ALAIN FONTEYNE*, PAULINE RICHEZ**, LILIANE MAROT**, LEILA BELKHIR***, DOMINIQUE TENNSTEDT** & JEAN-LUC GALA*§
*Center for Applied Molecular Technologies/DLD-Bio, Universite ́ catholique de Louvain, Brussels, **Department Dermatology, and ***Department of Infectious Disease, Saint-Luc University Hospital, Brussels, and §Defense Laboratories Department, ACOS Ops & Trg, Belgian Armed Forces
A 31-year-old patient presented with a diagnosis of granulomatous dermatophytosis based on the clinical aspect of the lesions and the rare presence of hyphae on direct microscopic examination of clinical material. A chronic evolution and progression of the disease, its resistance to a wide range of antifungal agents, the occasional presence of hyphae on direct examination but consistently negative cultures over a 5-year period prompted the use of amplification-based DNA analyses of several successive swab samples or skin biopsies. DNA was extracted using a combination of two semi-automated DNA isolation methods (FastPrep® preparation and NucliSENS® lysis magnetic extraction method). Identification relied both on sequence analysis of amplicons after SYBR® Green real-time PCR of the panfungal internal transcribed spacer 1 (ITS1) genetic target, as well as the unique amplicon melting curve profile of positive samples. Accordingly, Trichophyton rubrum was unambiguously identified in several clinical samples collected over a 7-month period. This case illustrates the contribution of DNA-based assays applied directly to sample biopsies for identifying causative agents in cases in which fungal pathogens are highly suspected but culture are repeatedly negative. It also pinpoints the benefit of combining semi-automated DNA preparation methods, analysis of ITS1 amplicon melting curve profiles and sequence analysis on repeated skin biopsy samples for unambiguous identification of the causative fungal species.
Keywords real-time PCR, internal transcribed spacer, granulomatous dermatophytosis, Trichophyton rubrum
____________
Introduction
For the last decade, DNA-based identification methods using amplification and DNA sequencing have been routinely used to better understand the taxonomy and phylogenetic diversity among fungi, in particular among the dermatophytes [1]. The DNA sequence data have been used to define molecular identification approaches for genotyping and identifying the dermatophytes in dermatological specimens, mainly nails and hair [2-6]. A reliable procedure for identifying pathogenic fungi in culture is based on PCR amplification of conserved genomic regions followed by sequence analysis of the amplicons [7]. In contrast, it appears far more challenging to obtain purified fungal DNA directly from clinical samples, especially when the fungal load is low as is often the case when fungal cultures from the same specimens remain negative, or when the patient is currently being or has recently been treated with antifungal agents. While several fungal DNA extraction protocols can be found in the literature, a promising approach is a bead-beating method followed by DNA purification [8]. Another potential issue when dealing directly with clinical samples that potentially contain fungi is the risk of false positive results due to sample contamination by ubiquitous fungal DNA in the environment. This can occur either directly during or after sample collection, or during the pre-analytical and analytical processing steps. If special attention is paid to avoid this problem, the random approach can be useful when clinical features and histology are suggestive of fungal infection but there is a lack microbiologic confirmation of the data. This is highlighted in the current case report. The patient initially presented with clinical symptoms which suggested nodular chronic granulomatous dermatophytosis on the left foot. The diagnosis relied on the clinical features and the rare presence of hyphae on direct microscopic examination of clinical material. Despite the progressive evolution of the disease over a few years, several attempts to confirm the diagnosis and identify the causative agent by culture failed. Nonetheless, hyphae could occasionally be found on direct examination. In this report, the DNA-based methodology used to successfully detect and identify the causative dermatophyte directly from biopsy and swab samples is detailed and discussed.
| Materials and methods
Case report
Patient (male, 31 years old) presented with a 5-year long history of dermatophytosis which was not responding to antifungal therapy. At presentation, the diagnosis of dermatophytosis was first suggested by the rare presence of hyphae in squamae on direct mycologic examination but culture remained consistently negative. Accordingly, the patient was initially treated on the basis of a presumed fungal infection with fluconazole 200 mg once a week for five months. However, since the patient did not respond, a second protocol was applied consisting of griseofulvine 500 mg a day which was replaced after five months with terbinafine 250 mg a day for 20 months. These antifungals did not prevent extension of the lesions. Therefore, at this time a pulse therapy involving itraconazole 200 mg twice a day for one week per month resulted in a significant improvement. However, after five months there was a reoccurrence of the lesions and treatment with itraconazole pulse and terbinafine 250 mg was begun. This combined therapy did not result in significant improvement and was replaced after 12 months by voriconazole 600 mg a day. While the cutaneous lesions regressed initially, they were still present 19 months after the initiation of this therapy. At this time, a periodic control of the biological parameters revealed that the patient was developing toxic hepatitis. Voriconazole was then replaced by pozaconazole 400 mg twice a day, but without major impact on cutaneous lesions. |
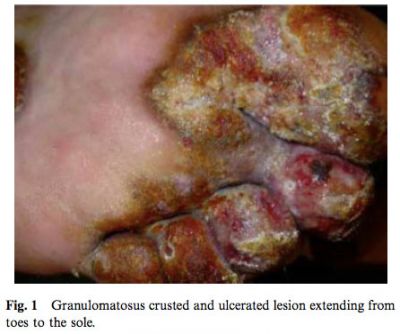
|
Despite continuous antifungal therapy, the dermatophytosis progressed in the following years, slowly invading both feet and one leg and leading with progressive increase in the number and size of the nodules (Fig. 1).
The hypothesis of primary immunodeficiency disorder as a cause for the progression of the disease was not supported by CD4 counts and the different types and subtypes of immunoglobulins. Swabs of the wound were repeatedly collected over a 5-year period to ascertain the initial diagnosis of dermatophytosis. While bacteria were occasionally recovered in cultures inoculated with these swabs and hyphae rarely noted on direct examination of skin scrapping, cultures for fungi remained consistently negative. In 2004, a biopsy was performed which provided histologic evidence compatible with the initial diagnosis of dermatophytic granuloma. In August 2007, the lack of favourable therapeutic outcome prompted investigations with alternative methods to confirm the initial diagnosis of dermatophytic granuloma. Molecular and histological examinations were carried out on successive cutaneous deep biopsies and swabs from a nodule. Samples were collected over seven months.
Microscopic observation and microbiological investigation
Skin scraps and swabs were examined microscopically for the presence of fungal elements using 10% KOH and a few drops of Parker’s ink. Attempts to cultivate the fungus were performed at 29°C and 37°C for 3 weeks on Sabouraud glucose agar without antibiotics.
DNA extraction
All DNA manipulations pre- and post-PCR were performed in separate designated rooms with separate pipetting devices to avoid contamination of the samples with foreign DNA. Furthermore, all equipment used in the preamplification steps was DNA-free and further irradiated by ultraviolet (UV) light to prevent contaminating DNA from causing false-positive results. Mas- ter-mixture water controls and DNA extraction controls were used for every batch of samples pro- cessed. The three sample biopsies processed for DNA extraction and amplification were taken from the same area on the left foot (Fig. 1). The samples were collected in the operating theatre and sent to the laboratory at room temperature in a sterile container without transport medium. They were stored at -20°C to be processed the next day. Comparable weight (about 20 mg) biopsy samples were minced with scalpels on a sterile glass slide and resuspended in 500 ml NucliSENS® lysis buffer (NucliSENS® lysis magnetic extraction reagents, NucliSENS® miniMAG System, Biomérieux bv, Boxtel NL) in a tube containing the FastPrep® Lysing Matrix A. The tubes were then shaken for 40 sec at 6 m/s in FastPrep®-24 MPTM homogenizer (FastPrep®, MP Biomedicals Europe sa, Brussels, BE) and then incubated at room temperature for 1 h. The cotton end of sterile swabs taken from the skin lesions were dropped in 1.5 ml Eppendorf tubes containing 500 ml NucliSENS® lysis buffer. Those tubes were then shaken vigorously and incubated at room temperature for 1 h. Total DNA was released from the homogenized suspension of biopsies and swabs using the NucliSENS® miniMAG system and reagents according to the manufacturer’s instructions. DNA was eluted in 50 ml. The whole DNA extraction process was performed in less than 1.5 h.
Control samples
Master-mixture water controls (n=50) were included as background control. In order to define a positive cut off for the ITS1 consensus real-time PCR assay, a panel of 18 aspirate or biopsies samples from patients with unrelated, non-fungal infections (bacterial prosthetic infections, n=9) or controls (osteoarthrosis, n=9) were processed as described above. The mean +/- standard deviation (SD) was calculated on all the control samples and their triplicate results. The cut-off value was defined as 2 SDs under the mean value obtained for the master-mixture water controls.
Fungal DNA detection and identification
A real-time PCR amplification of the internal transcribed spacer (ITS) 1 region was performed using previously described primers ITS1 5'-tccgtaggt- gaacctgcgg-3' and ITS2 5’-gctgcgttcttcatcgatgc-3’ [9]. The real-time PCR assay was performed using 5 ml of each DNA solution, 330 nM of each primer, 10 ml SYBR® Green I Master (Roche Diagnostics, Mannheim, Germany) in a total reaction volume of 20 ml. The reaction was initiated at 95°C for 5 min followed by 40 cycles of denaturation at 95°C for 10 sec, annealing at 60°C for 15 sec and extension at 72°C for 10 sec, followed by one cycle at 95°C for 5 sec, 65°C for 1 min, and 97°C with a continuous acquisition mode as requested for the analysis of the melting curves. Data were recorded as crossing points (Cps) on a Roche LightCycler® 480 System, using the analytical software LCS480 1.2.9.11 from the same manufacturer. Five ng of DNA from Aspergillus fumigatus strain DSM 63359 (Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH., Braunschweig, Germany) was used as a positive control.
All the amplification tests were performed in triplicate. The amplicons were sequenced on both strands on an automated 3130 Genetic Analyser apparatus (Applied Biosystems, Nieuwekerk, NL), using the Taq Dye Deoxy Terminator Cycle Sequencing kit from the same manufacturer. Sequences were compared against those in Genbank-EMBL using the blast algorithm available at http://www.ncbi.nlm.nih.gov/Blast.cgi. The whole process, from DNA amplification to sequence analysis, took less than 10 working hours spread on 1.5 day.
Results
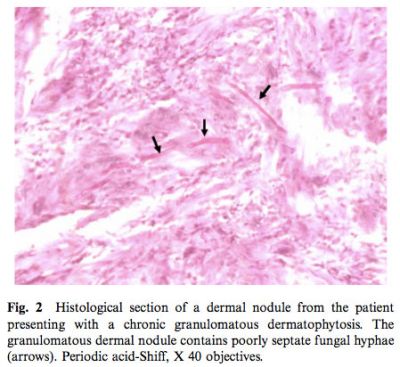
Fungal hyphae were observed in the same biopsy specimen used for molecular fungal detection and identification (Fig. 2).
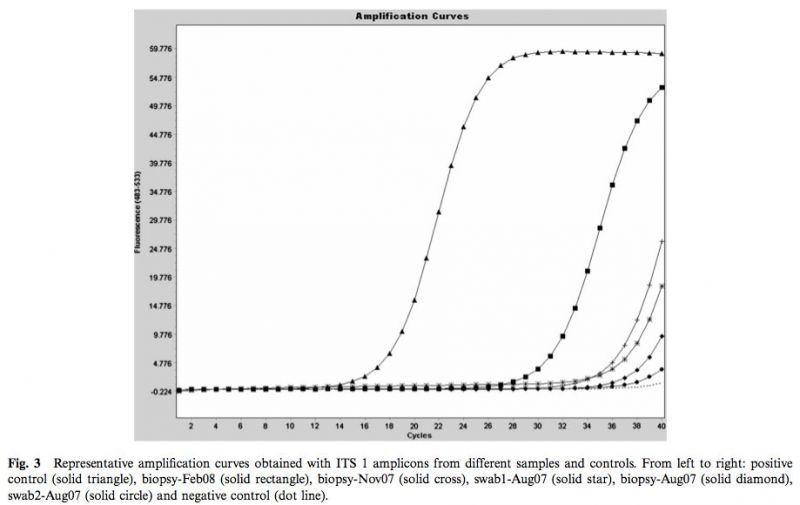
The Cp value obtained for 50 master-mixture water controls was 38.8±1.7 (mean +/- SD). The Cp value obtained from the 18 control biopsies or aspirates from patients with no fungal infections was 37.9±2.1 (mean +/- SD). Accordingly, a positive Cp value was defined as being less than the 35 cut off Cp value, corresponding to 2SD below the control Cp value. The real-time PCRs generated positive signals for the three biopsies and one of the two swab samples (Table 1).
 |
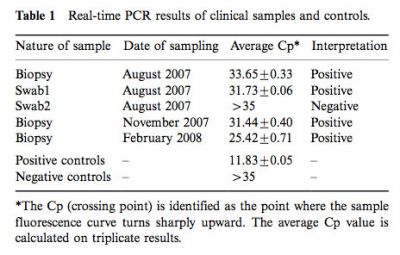 |
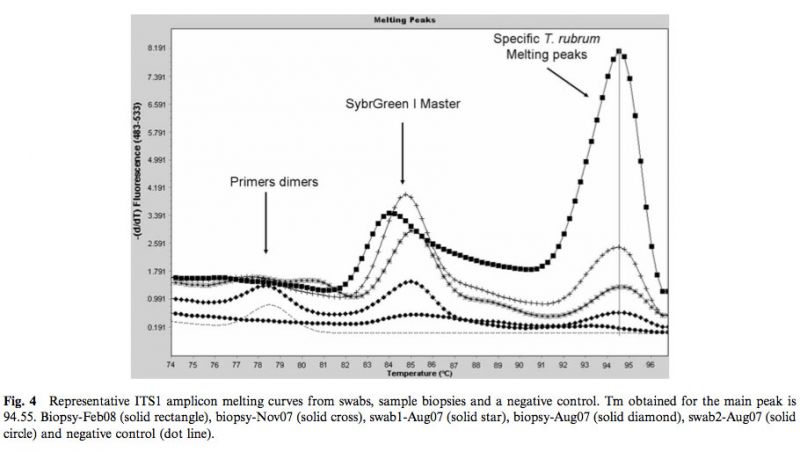
As shown in Fig.3, a marked progressive left shift of the amplification curves and Cps was observed with the successive clinical samples. The dissociation curves obtained with PCR products from each positive sample were highly similar, suggesting the presence of same fungal pathogen in these samples (Fig. 4). DNA sequence analyses of the 302-bp amplicons confirmed the sequence identity of the PCR product amplified from the four clinical samples. A comparison with Genbank-EMBL sequences showed that the query sequence was highly similar to a range of sequences from fungi related to Trichophyton rubrum (T. rubrum, T. raubitschekii, T. kanei and T. fischeri) [12]. The blast tree view derived from the multiple sequence alignment obtained at the NCBI showed that sequences published for isolates identified as Trichophyton violaceum, T. yaoundei, and T. soudanense are part of a distinct cluster.
Discussion and conclusion
The benefits of DNA-based amplification methods for unambiguous identification of the causative fungal agent are illustrated in this case of culture negative chronic granulomatous dermatophytosis. One of the reasons for consistently negative cultures over a 5-year period is most likely the administration of a wide range of antifungal agents which altered the ability of the fungus to be cultivated on nutrient media. The chronic evolution and progression of the disease despite chemotherapy with a range of antifungal agents, the occasional detection of hyphae in squamae but the persistent lack of fungal identification at the species level prompted DNA testing of a skin biopsy, as well as parallel collected swabs. The molecular detection and identification of T. rubrum in successive clinical samples collected over a 7-month period confirmed the initial diagnostic hypothesis based on the clinical aspects of the lesions and occasional detection of hyphae on direct microscopic examination. Identification of T. rubrum from tissue biopsies was also in concordance with histological evidence for the presence of hyphae in the same samples (Fig. 2). Although not proven, a progressive left shift of the amplification curves and Cps over time from successive skin biopsies of equal weight suggests a progressive increase in fungal load which was in line with the clinical evolution of the disease. Despite the lack of favorable outcome, an unambiguous identification of the pathogen allowed the confirmation of the presumed diagnosis. This confirmation was mandatory to justify maintaining an aggressive antifungal therapy which carried a risk of severe side-effects for the patient and highly expensive for the patient and healthcare system.
On a technical point of view, several issues were also addressed in this case. The first was to use a reliable fungal DNA isolation procedure due to the expected low fungal load in culture negative clinical samples. In accord with the recently published DNA extraction method to be directly used with clinical samples [8], it was decided to combine a DNA lysis buffer together with bead-beating for homogenization of the samples prior to semi-automated DNA purification. The FastPrep bead-beater and lysis buffer were first used to disrupt the tissue sample and obtain homogenized suspension. The NucliSENS® lysis magnetic extraction method, based on the chaotropic properties of Guanidinium salts and capture of DNA on magnetic silica beads, was then used to purify the total cellular DNA.
Compared to the reported DNA extraction procedure [8], this semi-automated DNA extraction method appeared easy and extremely fast (turn around time about 90 min vs. about 3 h) while having the potential for high throughput analysis without the risk of molecular carryover. It appeared to be highly effective, allowing successful fungal DNA recovery from the three successive skin biopsies and one of two successive swabs. To the best of our knowledge, this is the first report describing the use of combined rapid semi-automated methods for direct fungal DNA extraction from unfixed clinical samples.
A second issue was to sel ect the fungal genetic target. In order to screen our sample for a wide spectrum of dermatophytic and non dermatophytic species, it was decided to amplify the internal transcribed spacer region 1 rather than other specific fungal molecular targets. Indeed, sequencing ITS1 appears to be a reliable method to discriminate among most clinically relevant yeasts and moulds species [10,11]. In the current study, identification of T. rubrum relied on ITS1 sequence homology with T. rubrum sequences accessible in GenBank/EMBL database. In contrast, consistent ITS1 mismatches differenciated the T. rubrum cluster fr om another cluster which included T. soudanense and T. violaceum species. The resulting tree view was also in agreement with the molecular taxonomy of the T. rubrum complex, as previously published [12].
Thirdly, the SYBR® Green dye was preferred to a fluorescent probe for the real-time PCR assay. The former allowed comparing the amplicon melting curve profiles of successively processed clinical samples. Moreover, the value of amplicon melting curves as a screening test for identifying fungal DNA in clinical specimens was recently suggested [13].
However, SYBR® Green real-time amplification of ITS1 requires DNA sequencing for identifying fungi at the species level. In the current case, a species belonging to the T. rubrum group was consistently identified in clinical samples collected over more than half a year. It is of note that the species with ITS DNA sequences common to T. rubrum have been reduced to synonomy or reclassified within this species on the basis of phenotypic and molecular methods [12]. Results of the sequence analysis were in line with the diagnostic hypothesis of the dermatologist, the clinical features of the lesions, the histological evidence of dermal granuloma with hyphae in the same clinical samples, and the fact that T. rubrum is the most frequent cause of superficial as well as deep dermatophytosis [14].
A last issue was related to the specificity and sensitivity of our molecular approach. This was addressed at two different levels, i.e., the real-time PCR Cp results and interpretation of DNA sequence alignement. The panfungal primers used in our real-time PCR assay were published in 1990 and have been used ever since for amplifying DNA from fungal cultures, especially in the framework of taxonomical studies on clinically relevant fungi [9]. We are also routinely using them for genotyping fungal colony. The mean Cp value obtained from a panel of 18 control DNA samples confirmed the fungal specificity of our real-time PCR assay. However, these primers are not specific for the T. rubrum group. Samples contamination with another ITS-1 positive species would have led to co-amplifica- tion of both genetic targets and mixed amplicons. However, this was not seen on sequence analysis. In contrast, the presence of ITS1-negative contaminating fungus cannot be ruled out. It could also be argued that identification of T. rubrum in the biopsies resulted from skin colonization with this fungal species and not from an invasive infection. The consistent positive and negative results from patient and control samples respectively, as well as the weak PCR signal with only one of the two skin swabs make this hypothesis very unlikely.
Despite the advantages of DNA-based identification methods, the place of fungal genotyping in routine microbiological diagnosis is yet poorly defined. The costs of processing specimens as well as the need for skilled personal, sophisticated equipment, and well adapted infrastructure are clear limitations to a widescale use. While current technological constraints do not yet allow for widespread implementation of fungi genotyping in routine clinical mycology laboratories, those methods should be available within reference facilities. Indeed, as highlighted here, fungal genotyping has proved its ability to elucidate difficult cases (i.e., presumed fungal infection based on a suggestive histology but unconfirmed by culture) by bringing rapid and valuable DNA-based identification. In most cases of presumed dermatophytosis, the initial clinical diagnosis relying on the rare presence of fungal hyphae in skin samples would have been satisfactory. In the current case, the unusually long and bad evolution of the disease and its therapeutic antifungal resistance requested an unambiguous identification of the causative agent.
In conclusion, we report the case of a patient with a long-standing history of granulomatous dermatophytosis which was not responding to most antifungal agents. This case pinpoints the benefit of combining semiautomated DNA preparation methods, analysis of ITS1 amplicon melting curve profiles and sequence analysis on repeated skin biopsy samples for unambiguous identification of the causative fungal species, especially when microbiologic confirmation is lacking.
Acknowledgements
This work was supported by the Ministère de la Région wallonne - DGTRE Division de la Recherche et de la Coopération Scientifique (projects MYCOSERV no. 716572 and RESPIBAC no. 616313) and by the Department Management of Scientific & Technological Research of Defence (IRSD-RSTD; Royal High Institute for Defence) supporting research and develop- ment (grant MED-08).
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Gräser Y, De Hoog S, Summerbell RC. Dermatophytes: recognizing species of clonal fungi. Med Mycol 2006; 44: 199-209.
- Kardjeva V, Summerbell R, Kantardjiev T, et al. Forty-eight-hour diagnosis of onychomycosis with subtyping of Trichophyton rubrum strains. J Clin Microbiol 2006; 44: 1419-1427.
- Monod M, Bontems O, Zaugg C, et al. Fast and reliable PCR/ sequencing/RFLP assay for identification of fungi in onychomycoses. J Med Microbiol 2006; 55: 1211-216.
- Nagao K, Sugita T, Ouchi T, Nishikawa T. Identification of Trichophyton rubrum by nested PCR analysis from paraffin embedded specimen in trichophytia profunda acuta of the glabrous skin. Nippon Ishinkin Gakkai Zasshi 2005; 46: 129-132.
- Menotti J, Machouart M, Benderdouche M, et al. Polymerase chain reaction for diagnosis of dermatophyte and Scytalidium spp. onychomycosis. Br J Dermatol 2004; 151: 518-519.
- Turin L, Riva F, Galbiati G, Cainelli T. Fast, simple and highly sensitive double-rounded polymerase chain reaction assay to detect medically relevant fungi in dermatological specimens. Eur J Clin Invest 2000; 30: 511-518.
- Borman AM, Linton CJ, Miles SJ, Johnson EM. Molecular identification of pathogenic fungi. J Antimicrob Chemother 2008; 61(Suppl. 1): 7-12.
- Griffiths LJ, Anyim M, Doffman SR, et al. Comparison of DNA extraction methods for Aspergillus fumigatus using real-time PCR. J Med Microbiol 2006; 55: 1187-1191.
- White TJ, Burns T, Lee S, Taylor JW. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfland DH, Sninsky JJ, White TJ (eds). PCR Protocols: A Guide to Methods and Applications. New York: Academic Press, 1990: 315-332.
- Chen YC, Eisner JD, Kattar MM, et al. Polymorphic internal transcribed spacer region 1 DNA sequences identify medically important yeasts. J Clin Microbiol 2001; 39: 4042-4051.
- Iwen PC, Hinrichs SH, Rupp ME. Utilisation of the internal transcribed spacer regions as molecular targets to detect and identify human fungal pathogens. Med Mycol 2002; 40: 87-109.
- Gräser Y, Kuijpers AF, Presber W, de Hoog GS. Molecular taxonomy of the Trichophyton rubrum complex. J Clin Microbiol 2000; 38: 3329-3336.
- Schabereiter-Gurtner C, Selitsch B, Rotter ML, Hirschl AM, Willinger B. Development of novel real-time PCR assays for detection and differentiation of eleven medically important Aspergillus and Candida species in clinical specimens. J Clin Microbiol 2007; 45: 906-914.
- Chastain MA, Reed RJ, Pankey GA. Deep dermatophytosis: report of 2 cases and review of the literature. Cutis 2000; 67: 457-462.
Received 28 March 2008; Received in final revised form 18 June 2008; Accepted 26 July 2008
Correspondence: Pierre-Alain Fonteyne, Center for Applied Molecular Technologies, Université catholique de Louvain. Clos Chapelle-aux-Champs 30/46, 1200 Brussels, Belgium. Tel.: +32 2764 3157; Fax: +32 2764 3959; E-mail: pierre-alain.fonteyne@uclouvain.be
This paper was first published online on iFirst on 16 September 2008.


